
来源:德和衡律师
发布日期:2025年09月09日
编者按
随着全球医药行业的蓬勃发展和国际合作的不断深化,越来越多的医药企业将视野投向海外市场,为了医药企业能够更好地理解全球各地市场的法律环境、把握出海的法律要点、提前预判企业出海潜在风险并做好相应的准备,天牧生命科学法律团队特编写“药械大健康企业出海法律指南”系列。
目录
1.引言
2.药械大健康企业出海美国的支持性因素
2.1规模庞大且增长迅速的市场现状为药企出海提供更多可能性
2.2美国政府对于药械大健康行业出台一系列支持性政策和举动
3.美国药械大健康市场的运作模式概述
3.1市场构成
3.2市场体系
4.美国药械大健康市场的法律监管体系
4.1法律监管机构汇总
4.2药械大健康市场相关法律框架体系
5.美国药械大健康市场准入要点
5.1药品准入类型
5.2医疗器械准入流程
6.美国药械大健康市场的采购体系
6.1药品采购体系
6.2医疗器械采购体系
6.3采购中的返利与回扣机制
6.4回扣机制的法律监管体系
7.美国药械大健康市场的销售体系
7.1药品销售的市场结构
7.2医保报销
7.3销售流程
8.结语
0 1
引言
随着全球经济一体化的深入发展,国际医药产业迎来了前所未有的变革时期。在这一浪潮中,越来越多的药械大健康企业将目光投向海外,寻求更广阔的市场空间与发展机遇。美国,凭借其规模庞大且增长迅速的医药市场、持续创新的科技实力以及完善的产业支持体系,成为众多企业出海布局的重要目标。
美国药械大健康市场不仅拥有高额的消费需求,在技术研发和市场运作方面也处于世界领先地位,为企业提供了丰富的发展可能性。然而,进入美国市场并非坦途,其背后复杂的法律体系和严格的监管要求,成为企业必须跨越的重要障碍。从产品准入的审批流程,到市场运营的各个环节,都受到多层次法律规范的约束。如果企业对这些法律要点缺乏深入了解,可能会面临产品无法进入市场、运营违规受罚等风险,给企业带来巨大损失。
基于此,本指南聚焦美国药械大健康市场,深入剖析其法律监管体系、市场准入规则、采购与销售环节的法律规范等核心内容,旨在为准备出海美国的药械大健康企业提供全面、精准且实用的法律指引,助力企业在复杂的美国市场中稳步前行,有效规避法律风险,实现商业目标。
02
药械大健康企业出海美国的支持性因素
2.1
规模庞大且增长迅速的市场现状为药企出海提供更多可能性
美国是全球最大的医药市场,占据全球医药市场的主导地位。截止2024年年底,全球药品市场规模约为1.61万亿美元,美国占据重要份额。美国医药市场呈现出持续增长的态势。根据最新的数据表明,2024年美国医药市场的总额约为4059亿美元,复合年增长率(CAGR)约为3.2%,其增长的动力主要包括技术创新、人口老龄化、慢性疾病负担加重以及中产阶级人口的增长。此外,核药市场的扩容也为整体市场增长提供了新的动力。
在市场结构方面,美国医药市场涵盖了制药、生物技术、医疗设备、诊断服务等多个细分领域。其中,制药和生物技术是美国医药市场的核心板块,创新药和生物制品是其市场增长的主要驱动力。值得一提的是,美国的核药市场也在快速发展,在2024年这一年中美国FDA就已批准50款新药。美国医药市场参与者众多,包括大型制药公司、生物技术企业以及众多创新型初创公司等。
与其他国家相比,美国医药市场具有以下特点:一是创新药价格高,仿制药价格低,创新药价格为全球平均水平的4.2倍,而仿制药仅为全球平均水平的0.7倍。二是药品定价自由化,药企与医药批发零售商、PBM(药品福利管理)、保险公司等机构分别谈判,利用规模效应降低价格。三是研发和创新能力突出,美国在基因治疗、细胞疗法等前沿技术领域处于领先地位。四是市场集中度较高,例如核药房供应的SPECT诊断用药市场集中度达到99%。
2.2
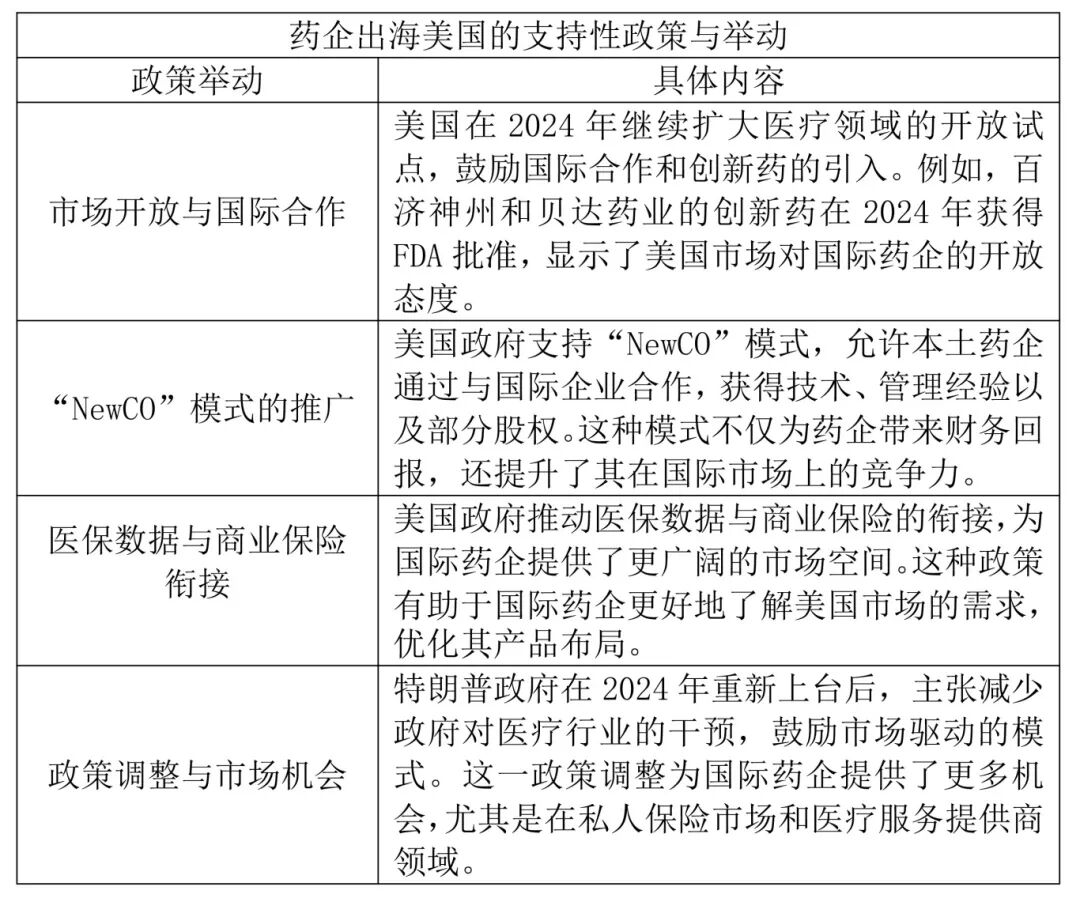
美国政府对于药械大健康行业出台一系列支持性政策和举动
美国的医药政策不仅推动了本国医药市场的发展,也为国际医药企业提供了出海机会。通过《通胀削减法案》和《平价医疗法案》的实施,美国政府在控制医疗费用的同时,也为医药市场创造了更广阔的需求空间。FDA的灵活审评审批政策和供应链回流政策进一步增强了美国医药产业的竞争力。此外,美国政府通过市场开放和国际合作,为国际药企提供了更多进入美国市场的机会。
03
美国药械大健康市场的运作模式概述
3.1
市场构成
美国医药市场由多个支付方组成,主要包括政府支付方市场 (Government Payer Market) 和商业保险支付方市场 (Commercial Payer Market)。
3.1.1政府支付方市场 (Government Payer Market)
3.1.1.1 Medicare: 由联邦政府运营,主要覆盖65岁及以上人群,以及部分65岁以下的残疾人和终末期肾病患者。其中Part B和Part D是与医药市场紧密相关的两个重要组成部分。

3.1.1.2 Medicaid: 由联邦政府和各州共同出资,覆盖符合条件的低收入人群。
3.1.2商业保险支付方市场 (Commercial Payer Market)
3.1.2.1 Medicare Advantage(Part C): 由商业保险公司承办的Medicare补充计划,涵盖Medicare的全部或部分福利,并可能提供额外服务。
3.1.2.2 雇主健康保险 (Employer Health Insurance): 由雇主为员工购买的商业保险,通常由商业保险公司承办。
3.2
市场体系
美国医药市场是一个复杂且高度发达的体系,主要包括药品和医疗器械的采购、销售渠道、监管框架以及市场准入机制。其运作模式高度依赖于集中采购组织(GPO)、药品福利管理机构(PBM)、保险公司、药房和医疗器械制造商等多方参与。
04
美国药械大健康市场的法律监管体系
4.1
法律监管机构汇总
4.1.1美国食品药品监督管理局(FDA)
4.1.1.1概述
美国食品药品监督管理局(FDA)是美国卫生与公众服务部(HHS)下属的联邦政府机构,负责监管食品、药品、医疗器械、化妆品、兽药、烟草制品等产品的安全性、有效性和质量。FDA的使命是保护和促进公众健康,确保这些产品在使用或消费过程中不会对公众健康造成威胁。
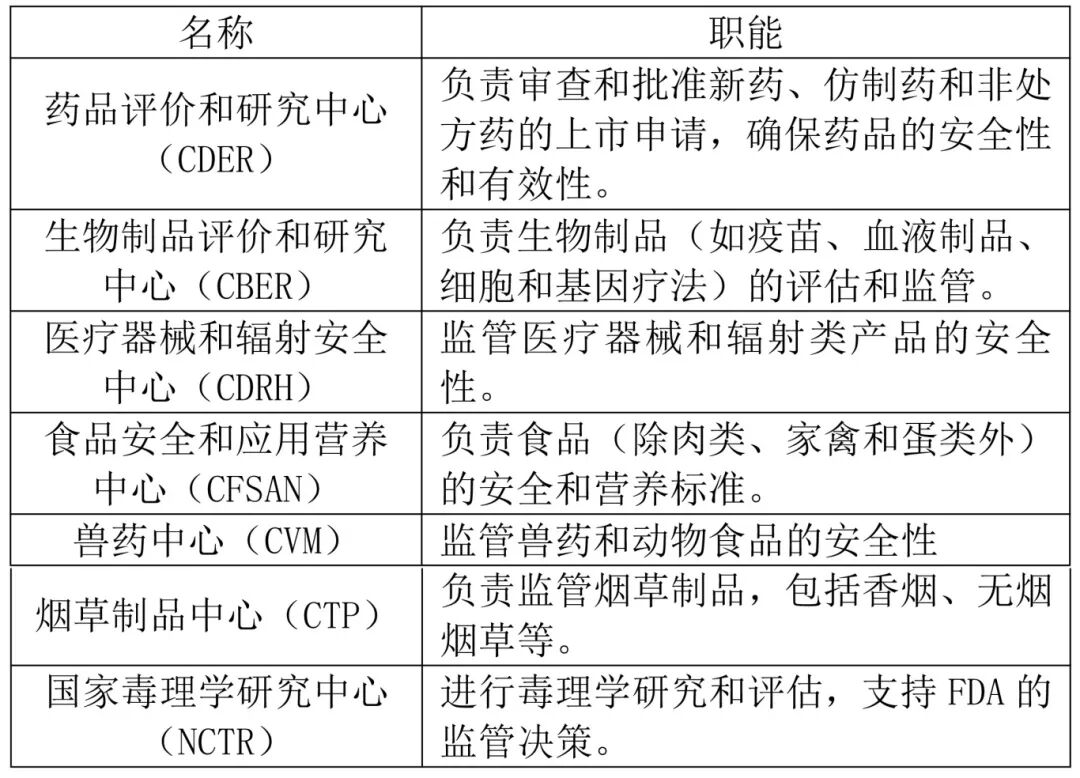
4.1.1.2组织架构
4.1.1.3监管范围
4.1.1.3.1食品:包括膳食补充剂、瓶装水、食品添加剂等。
4.1.1.3.2药品:包括处方药、非处方药、生物制品。
4.1.1.3.3医疗器械:包括诊断设备、治疗设备、手术植入物等。
4.1.1.3.4化妆品:包括皮肤护理、指甲油、香水等。
4.1.1.3.5兽药和动物食品:包括宠物食品、兽药。
4.1.1.3.6烟草制品:包括香烟、无烟烟草等。
4.1.1.3.7辐射产品:如微波炉、X射线设备等。
4.1.2医疗保险和医疗补助服务中心(CMS)
4.1.2.1概述
美国医疗保险和医疗补助服务中心 (Centers for Medicare & Medicaid Services,简称CMS) 是美国卫生与公众服务部(HHS)下属的一个联邦机构,主要负责管理美国的主要公共健康保险计划。
4.1.2.2主要职责
4.1.2.2.1管理联邦健康保险计划:
※Medicare( 联邦医疗保险):为65岁及以上老年人、65岁以下的残疾人以及患有终末期肾病的患者提供医疗保险。
※ Medicaid( 医疗补助):为低收入人群提供医疗保障,由联邦政府和各州共同资助。
※Children's Health Insurance Program(CHIP, 儿童健康保险计划):为无法负担私人医疗保险的儿童提供医疗保障。
※ Health Insurance Marketplace( 健康保险市场):通过《平价医疗法案》 (Affordable Care Act) 管理联邦和州的健康保险市场。
4.1.2.2.2制定和执行医保政策:
※CMS负责制定 Medicare和Medicaid的 报销政策,决定哪些医疗服务和药品可以报销。
※监督医疗服务质量,确保长期护理机构和临床实验室符合标准。
4.1.2.2.3数据收集与分析:
※CMS收集和分析医疗保健数据,发布研究报告,帮助制定政策和改进医疗保健系统。
4.1.2.2.4反欺诈与滥用:
※CMS致力于打击医疗保健领域的欺诈和滥用行为,保护医保资金的安全。
4.1.2.2.5推动医疗改革:
※CMS致力于通过简化行政流程、推广电子健康记录等方式,提升医疗保健系统的效率和质量。
4.1.3其他监管机构
4.1.3.1美国国会
通过立法确立医药监管框架,如《联邦食品、药品和化妆品法案》,并监督FDA的执行情况。
4.1.3.2州政府卫生局
各州卫生局也设有药政机构,负责地方层面的药品和医疗器械监管。
4.2
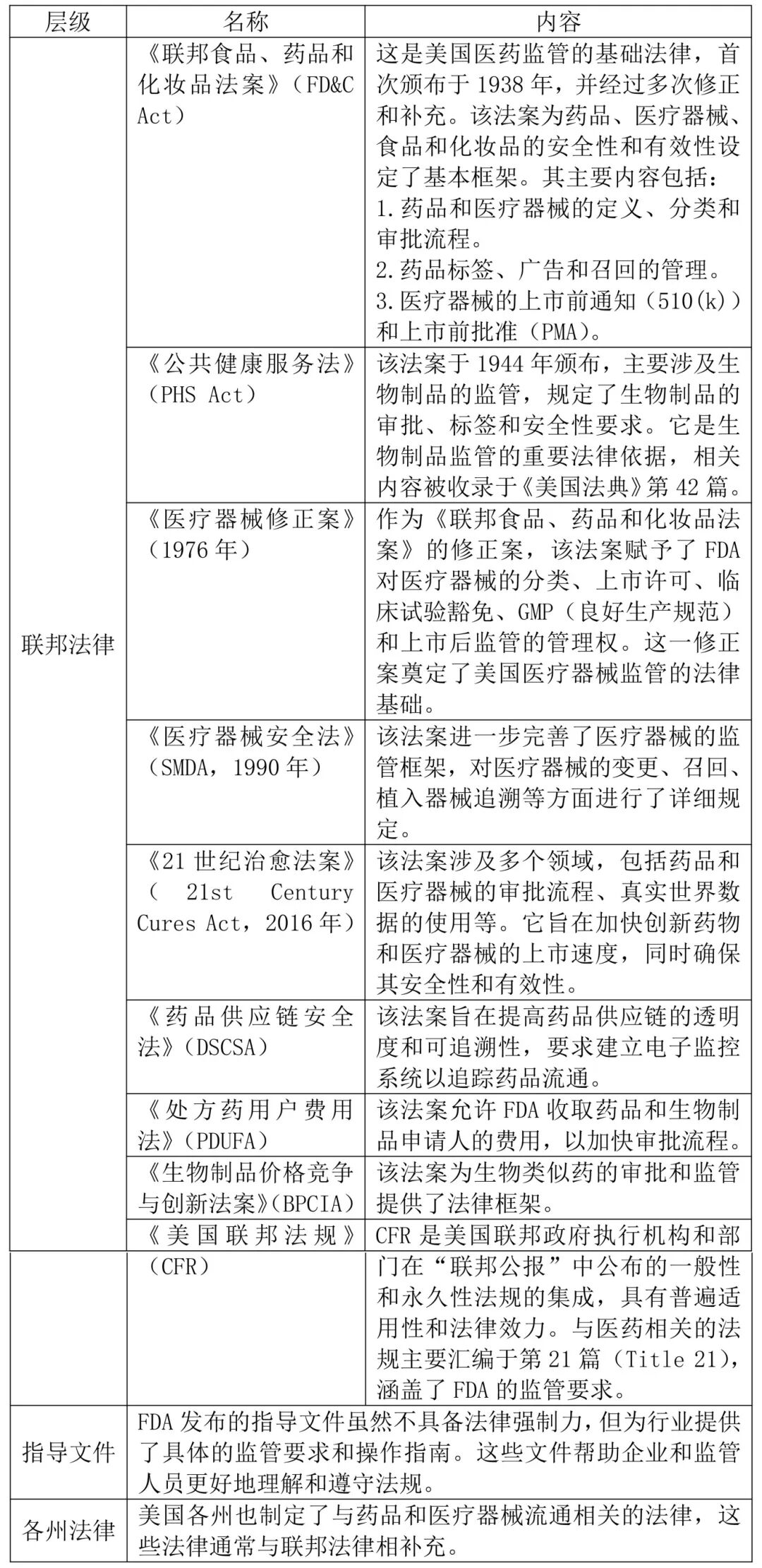
药械大健康市场相关法律框架体系
05
美国药械大健康市场准入要点
5.1
药品准入类型 [1]
5.1.1研究性新药(IND)
5.1.1.1IND申请的定义与目的
研究性新药申请 (Investigational New Drug Application,IND) 是临床研究申办者向美国食品药品监督管理局(FDA)提交的申请,旨在获得对人体使用研究性药物或生物制品的授权。IND申请是新药研发过程中进入人体临床试验的关键步骤,其目的是确保药物在进入人体试验前具有足够的安全性。
5.1.1.2IND申请的类型
※商业IND:由制药公司或研究机构提交,用于支持新药的临床开发。
※研究者IND:由医生或研究者自行提交,用于开展特定的临床研究,例如探索药物的新适应症或患者群体。
※紧急使用IND:在紧急情况下,允许FDA授权使用实验性药物,适用于没有足够时间按常规流程提交IND的情况。
※治疗性IND:针对严重或危及生命的疾病,允许在临床试验之外使用实验性药物。
5.1.1.3IND申请的关键组成部分
5.1.1.3.1临床前数据:
※包括动物药理学和毒理学研究数据,用于评估药物在人体初步试验中的安全性。
※还需提供药物在人体应用的任何经验(如国外使用数据)。
5.1.1.3.2生产信息:
※包括药物成分、生产方法、稳定性以及质量控制措施。
※确保企业能够持续生产质量一致的药物批次。
5.1.1.3.3临床试验方案及研究者信息:
※提供详细的临床试验方案,包括试验设计、目标、受试者选择标准等。
※提供研究者资质信息,确保其具备开展临床试验的能力。
※包括知情同意书、机构审查委员会(IRB)的审查意见以及遵守IND法规的承诺。
5.1.1.4IND申请的提交与审批流程
5.1.1.4.1提交准备:
申办者需整理所有必要信息和文件,包括临床前数据、临床试验方案、生产信息和安全监测计划。
商业IND需以eCTD格式通过电子提交网关(ESG)提交,研究/非商业IND可选择电子提交。
5.1.1.4.2审批流程:
提交IND申请后,申办者需等待30个日历日才能启动临床试验。
在此期间,FDA将对申请进行安全性审查,以确保受试者不会面临不合理的风险。
FDA可能会要求补充材料或提出问题,申办者需及时响应。
5.1.1.5IND申请的后续管理
5.1.1.5.1持续报告:
※在临床试验期间,申办者需定期向FDA提交进展报告,包括试验结果、安全性数据等。
※如遇重大问题或变更,需及时通知FDA。
5.1.1.5.2扩大准入:
※在某些情况下,FDA允许通过IND申请扩大实验性药物的使用范围,例如在临床试验之外用于严重疾病患者。
5.1.2新药申请(NDA)
5.1.2.1NDA的定义与目的
NDA是用于批准新药(包括化学药品和生物制品)在美国上市的正式申请。其目的是确保药品的安全性、有效性和质量。
5.1.2.2NDA的申请流程
5.1.2.2.1临床前研究
在提交NDA之前,药企需要完成临床前研究,包括实验室和动物模型中的安全性评估、药理学研究等,以确定药物的作用机制和初步安全性。
5.1.2.2.2临床试验申请(IND)
※在启动临床试验之前,药企需向FDA提交临床试验申请(IND),包含临床前研究数据、研究方案、研究者信息等。
※IND获得批准后,方可开展临床试验。
5.1.2.2.3临床试验
※I期:主要研究药物的安全性和耐受性,通常招募少量健康志愿者。
※II期:评估药物的有效性和安全性,招募一定数量的患者。
※III期:进一步验证药物的有效性和安全性,招募大量患者,为NDA提供关键数据。
5.1.2.2.4提交NDA
※完成临床试验后,药企需整理所有研究数据,向FDA提交NDA。
※NDA需包含以下内容:药物的研发背景、化学结构、生产工艺、质量控制。临床前和临床试验的详细数据。
5.1.2.2.5FDA审核
※FDA对NDA进行全面审核,包括药学审评、医学审评、统计审评等。
※审核过程中,FDA可能会要求补充材料或召开听证会。
5.1.2.2.6批准上市
※若FDA认为药物的益处大于风险,将批准新药上市。
※企业获得上市许可后,可将新药推向市场。
5.1.2.3NDA的审批类型
※优先审评 (Priority Review): 针对突破性疗法或未满足临床需求的药物,审批时间缩短。
※加速批准 (Accelerated Approval): 基于替代终点提前批准,需后续验证临床获益。
5.1.3简化新药申请(ANDA)
5.1.3.1ANDA的定义与目的
ANDA(Abbreviated New Drug Application, 简化新药申请)是美国FDA批准仿制药上市的专门申请程序。ANDA适用于专利期已过或专利被撤销的原研药的仿制药,旨在证明仿制药与原研药在安全性、有效性和质量上具有等同性。
5.1.3.2ANDA的主要组成部分
※参考上市药物 (Reference Listed Drug, RLD): 仿制药必须参照已获FDA批准的原研药。
※化学、制造和控制 (CMC): 包括药物成分、生产工艺和质量控制。
※生物等效性 (Bioequivalence): 证明仿制药与原研药在人体内的吸收和利用程度相同。
※标签和包装:必须与原研药一致。
※行政文件:包括完整的申请表格和相关证明材料。
5.1.3.3ANDA的申请条件
※拟申请ANDA的药品必须与RLD具有相同的活性成分、剂型、规格、给药途径、适应症。
※必须证明与RLD生物等效。
※生产过程需符合美国联邦管理法21 CFR的cGMP(动态药品生产质量管理规范)。
5.1.3.4ANDA的申请流程
5.1.3.4.1前期准备:
※申请FEI号 (FDA企业识别号)、DUNS 号(数据通用编号系统)。
※指定美国代理人(US Agent),负责与FDA沟通。
※申请ANDA号和ESG号(电子提交网关账号)。
※准备并支付GDUFA费用(仿制药使用者费用)。
5.1.3.4.2提交申请:
※按照eCTD格式准备和提交ANDA申请。
※FDA在收到申请后60天内进行初步审查,确认资料完整性。
5.1.3.4.3审评过程:
※FDA进行化学、微生物、生物等效性和标签审评。
※可能会发出信息请求函(IR)或学科审批函(DRL),要求补充信息。
5.1.3.4.4现场检查:
※FDA可能对生产场地进行cGMP符合性检查。
5.1.3.4.5最终决定:
※如果审评通过,FDA将批准ANDA申请,允许仿制药上市。
5.1.3.5ANDA的优势
※简化流程:ANDA不要求提供完整的临床前和临床试验数据,只需证明生物等效性。
※成本效益:相比新药申请(NDA),ANDA的开发和审批成本较低。
※市场准入:为仿制药提供了快速进入美国市场的路径,有助于降低药品价格。
5.1.3.6注意事项
※生物等效性研究:ANDA申请的核心是证明仿制药与原研药的生物等效性,需严格按照FDA指南进行。
※标签一致性:仿制药的标签必须与RLD一致,除非FDA允许的差异。
※费用支付:ANDA申请人需支付GDUFA费用,包括审评费、项目费和设施费。
5.1.4治疗性生物制剂应用(BLA)
5.1.4.1BLA的定义
BLA(Biologics License Application, 生物制品许可申请)是向美国食品药品监督管理局(FDA)提交的申请文件,用于支持评审和最终批准生物制品在美国上市和销售。生物制品包括疫苗、血浆制品、重组蛋白、单克隆抗体等,通常用于治疗癌症、自身免疫性疾病和其他严重疾病。
5.1.4.2BLA的申请内容
※生产工艺:详细描述生物制品的生产过程,包括细胞系、培养基、纯化步骤等。
※化学和药理学数据:提供生物制品的化学结构、纯度、稳定性以及药理学特性。
※临床数据:包括临床试验的设计、受试者数据、安全性评估和疗效结果。
※质量控制:确保生物制品在生产过程中符合FDA的质量标准(cGMP)。
※标签和包装:提供产品的标签和包装信息,确保符合FDA要求。
5.1.4.3BLA的申请流程
5.1.4.3.1前期准备:
※企业需完成临床前研究和临床试验,确保生物制品的安全性和有效性。
※准备完整的申请文件,包括所有必要的数据和报告。
5.1.4.3.2提交申请:
※企业需通过FDA的电子提交系统(eCTD)提交BLA申请。
※FDA在收到申请后60天内进行初步审查,确认资料完整性。
5.1.4.3.3审评过程:
※FDA对申请文件进行详细审评,包括化学、药理学、临床数据和质量控制。
※如需补充信息,FDA会发出信息请求函(IR)或学科审批函(DRL),企业需在规定时间内回复。
5.1.4.3.4现场检查:
※FDA可能对生产场地进行cGMP符合性检查,确保生产过程符合质量标准。
5.1.4.3.5最终决定:
※如果审评通过,FDA将批准BLA申请,并颁发生物制品许可证。
5.1.4.4应用领域
※癌症治疗:如单克隆抗体、CAR-T细胞疗法等。
※自身免疫性疾病:如类风湿性关节炎、强直性脊柱炎等。
※传染病:如疫苗和抗病毒药物。
5.1.4.5注意事项
※数据完整性:BLA申请需提供完整的临床和非临床数据,任何缺失或不完整的信息都可能导致申请被拒。
※质量控制:生物制品的生产过程需严格符合cGMP要求,确保产品质量和安全性。
※费用支付:企业需支付相应的申请费用,包括审评费和设施检查费。
5.1.5非处方药(OTC)的药物申请
5.1.5.1OTC药物的定义
非处方药 (OTC,Over-the-Counter) 是指不需要医生处方即可购买的药品。在美国,OTC药物涵盖从痤疮治疗到体重控制等多个品类,目前市场上销售的OTC药物超过30万种。
5.1.5.2OTC药物的申请途径
5.1.5.2.1新药申请(NDA):
如果OTC药物的活性成分的安全性和有效性尚未被FDA认可,则需要通过完整的NDA流程,提交临床试验数据以证明其安全性和有效性。
5.1.5.2.2OTC专论 (OTC Monograph):
如果OTC药物的活性成分已被FDA认为是“一般认为安全有效”(GRASE),则可以通过OTC专论路径直接上市,无需预批准。
5.1.5.3OTC专论 (OTC Monograph)
OTC专论是FDA对OTC药物活性成分的安全性和有效性进行统一审查后发布的标准文件。如果OTC药物符合专论中的要求,可以直接上市,但需满足以下条件:
※活性成分必须符合专论要求。
※生产商需进行FDA厂址登记,并指定美国代理人。
※申请国家药品编号(NDC)。
※产品需在FDA进行列名,并每年更新两次。
※标签必须符合FDA规定。
※生产工厂需符合动态药品生产管理规范(cGMP)。
5.1.5.4OTC药物的注册流程
※获取DUNS号码:DUNS(数据通用编号系统)是一个9位数字的全球编码系统,用于企业识别。
※企业注册:OTC药物的生产商、重新包装商或进口商需在FDA进行企业注册。
※申请NDC标签代码:为OTC药物申请一个独一无二的国家药品编号(NDC)。
※提交结构化产品标签(SPL):将NDC标签代码转换为结构化产品标签(SPL)格式的XML文件,并提交给FDA。
※产品列名:将所有OTC药物的详细信息列出,并在FDA进行列名,包括产品标识、说明、标签等。
5.1.6DMF注册
5.1.6.1DMF的定义
DMF(Drug Master File, 药物主文件)是提交给美国食品药品监督管理局(FDA)的保密文件,用于提供药品生产、加工、包装和储存过程中使用的设施、流程和物品的详细信息。DMF的主要目的是保护生产商的商业机密,同时为FDA审评药品提供关键数据支持。
5.1.6.2DMF的类型
※Type I:已废止。
※Type II:涉及活性药物成分(API)、原料药中间体、用于制备药物的材料和药品的信息。
※Type III:聚焦包装材料,如药品包装容器等。
※Type IV:涉及辅料、着色剂、香料及其原料等。
※Type V:用于提交其他可被FDA接受的信息,如特定工艺或技术描述。
5.1.6.3注册流程
5.1.6.3.1资料准备
※DMF文件需按照ICH M4的CTD格式整理,并以eCTD格式提交(Type I除外)。
※所有文档需为英文版本,非英文部分需翻译。
※文件内容包括化学、制造和控制(CMC)信息、生产工艺、稳定性数据等。
5.1.6.3.2资料提交
※提交前需获取DMF号(预分配号)。
※文件小于10G时,通过FDA的电子提交网关(ESG)提交;超过10G时,可通过ESG或物理媒介 (如CD-ROM)提 交。
※提交时需选择正确的FDA中心 (如CDER或CBER), 若涉及多个中心需提前咨询。
5.1.6.3.3FDA审评
※行政审评:通常在提交后2-3周内完成。
※完整性审评:对于原料药 (Type II DMF), 需缴纳GDUFA费用后进行,通常需要60天。
※技术审评:当DMF被 IND、NDA、ANDA或BLA引用时,FDA 才会进行技术审评。
5.1.6.3.4审评结果
※DMF没有“批准”或“不批准”的说法。对于 Type II DMF, 通过技术审评后,FDA会颁发 “First Adequate Letter”或“No Further Comment Letter”。
5.1.6.4DMF的激活与引用
※DMF持有人需激活DMF编号,并通知相关药品申请人,以便在提交NDA或ANDA时引用。
※持有人需定期更新DMF文件,特别是在生产或质量控制过程发生变化时。
5.1.6.5合规性与检查
※DMF的审评过程涉及生产设施的cGMP合规性检查。
※FDA可能会对生产设施进行现场检查,以确认其符合相关生产和质量标准。
5.2
医疗器械准入流程 [2]
5.2.1确定产品分类
FDA根据风险等级将医疗器械分为三类:
※Class I(低风险):如医用手套、压舌板等。大多数Class I器械只需进行企业注册和产品列名,通常豁免上市前通知。
※Class II(中风险):如注射器、激光美容仪等。Class II器械通常需要提交510(k)上市前通知。
※Class III(高风险):如植入式心脏起搏器、人工心脏瓣膜等。Class III器械需要提交PMA(上市前批准)申请。
5.2.2企业注册与产品列明
所有医疗器械企业(包括国外企业)在向美国市场销售产品前,必须完成以下步骤:
※企业注册:企业需在FDA进行注册,并每年更新注册信息。
※产品列名:企业需向FDA提交产品信息进行列名,这是产品进入美国市场的必要步骤。
5.2.3提交上市前申请
5.2.3.1510(k)(上市前通知)
5.1.2.3.1定义与目的
510(k)上市前通知 (Premarket Notification) 是医疗器械制造商向美国食品药品监督管理局(FDA)提交的申请文件,目的是证明即将上市的医疗器械与已合法上市的同类器械(称为“predicate device”)在安全性和有效性方面具有实质等同性(Substantially Equivalent, SE)。
5.1.2.3.2适用范围
510(k)适用于大多数Class II(中等风险)医疗器械,以及部分Class I和Class III器械。Class I器械通常豁免510(k)要求,但需满足企业注册和产品列名要求。
5.1.2.3.3510(k)的类型
※传统510(k):适用于首次上市的器械或对已批准器械的重大变更。
※特殊510(k):适用于对现有器械的细微设计或标签变更,需提供风险分析或摘要报告。
※简略510(k):适用于符合FDA指导文件或自愿共识标准的器械。
5.1.2.3.4提交内容
※申请函:包括企业基本信息、产品名称、型号、分类及预期用途。
※目录:列出所有提交文件。
※产品描述:包括设计、材料、性能、标签和使用说明。
※实质等同性比较:将新器械与 predicate device 进行对比,证明其在预期用途、设计、材料、性能、安全性和有效性方面的等同性。
※测试数据:包括生物相容性测试、灭菌验证、有效期验证和性能测试。
※临床数据(如有):支持器械的安全性和有效性。
5.1.2.3.5审批流程
※提交:通过FDA的eCopy系统提交电子版510(k)文件。
※初步审查:FDA在90天内完成技术审查,可能要求补充信息。
※批准:如果器械被认定为实质等同,FDA将发出批准函,允许产品上市。
5.1.2.3.6注意事项
美国代理人:国外企业需指定美国代理人,协助FDA沟通和现场检查。
持续合规:获得510(k)批准后,企业需遵守FDA的质量体系要求(如cGMP)和上市后监管要求。
5.2.3.2PMA(上市前批准)
5.2.3.2.1定义
Premarket Approval(PMA, 上市前批准)是美国FDA对III类医疗器械(高风险医疗器械)进行科学和法规审查的严格审批程序,旨在评估这些器械的安全性和有效性。III类器械通常包括那些用于支持或维持生命、对预防人类健康损害至关重要的设备,例如人工心脏瓣膜、心脏起搏器等。
5.2.3.2.2PMA的适用范围
※大部分III类医疗器械。
※少部分II类医疗器械,特别是那些风险较高或缺乏合法上市同类器械的产品。
5.2.3.2.3PMA的申报方式
※传统PMA:一次性提交完整的申请文件,包括非临床和临床研究数据。
※模块化PMA:允许申请人在不同时间提交各个模块(如非临床数据、制造信息等),同时继续收集和分析临床数据。这种方法可提高审查效率。
※人道主义器械豁免(HDE):适用于针对罕见疾病或状况的医疗器械。
5.2.3.2.4PMA的审评流程
※接受性审核:FDA对提交的申请文件进行初步审查,以确定其完整性。
※实质性审评:FDA对申请文件进行深入的科学、法规和质量管理体系审查。
※专家组审评:对于复杂的申请,FDA可能召集外部专家进行咨询。
※最终决定:FDA将在收到申请后的180天内完成审查并做出决定。可能的结果包括批准、可批准信、不可批准信或拒绝批准。
5.2.3.2.5PMA申请的关键要求
※临床数据:申请人需提供充分的临床试验数据,以证明器械在预期用途下的安全性和有效性。
※非临床数据:包括设计验证、生物相容性测试、灭菌验证等。
※质量体系:申请人需证明其生产过程符合FDA的质量管理体系要求。
5.2.3.2.6PMA与510(k)的区别
※适用范围:PMA适用于III类医疗器械,而510(k)适用于II类医疗器械。
※审查严格性:PMA是FDA最严格的审批程序,要求提供完整的临床和非临床数据;510(k)则要求证明器械与已上市器械的实质等同性。
※审批时间:PMA的审批时间通常比510(k)长,可能需要1-2年。
5.2.3.3De Novo 分类请求
5.2.3.3.1定义
De Novo 分类请求 (De Novo Classification Request) 是美国FDA为新型或创新医疗器械提供的分类申请程序,适用于那些无法通过传统510(k)路径获得市场准入的器械。这些器械通常因为缺乏合法销售的同类产品 (predicate device), 无法证明“实质等同性” (Substantially Equivalent),从 而需要通过De Novo程序进行分类。
5.2.3.3.2适用范围
※没有合法销售的器械可用于510(k)路径中的“实质等同性”判定。
※器械被自动归为III类,但其风险可通过一般控制或特殊控制措施合理保证安全性和有效性。
※器械属于低到中等风险,且不适合通过PMA(上市前批准)路径。
5.2.3.3.3提交De Novo请求的两种途径
※收到510(k)的非实质性等同(NSE)通知后:如果企业在510(k)申请中收到“非实质性等同”(NSE)通知,可在30天内提交De Novo请求。
※直接提交De Novo请求:当企业确定没有合法销售的器械可用于“实质等同性”判定时,可以直接提交De Novo请求。
5.2.3.3.4De Novo请求的内容
※封面需标注“De Novo分类请求”。
※器械描述,包括技术特征、预期用途、附件和组件。
※建议的分类及支持数据,包括非临床和临床数据(如适用)。
※特殊控制措施,用于合理保证器械的安全性和有效性。
※器械可能带来的益处与风险的描述。
5.2.3.3.5审查流程
※验收审查:FDA在收到De Novo请求后,会进行行政审查,评估申请的完整性。
※实质性审查:FDA将评估器械的安全性和有效性,确定其是否可通过一般控制或特殊控制措施合理保证安全有效。
※附加信息:如果需要,FDA会发送附加信息函,要求申请者在180天内提供完整回复。
※最终决定:如果 De Novo 请求获批,器械将被分类为I类或II类,并可上市销售。
5.2.3.3.6De Novo请求的优势
※市场准入:为新型器械提供了一条相对快速的市场准入路径。
※避免PMA:避免了更复杂和耗时的PMA流程。
※作为未来510(k)的比对产品:获批的器械可作为未来510(k)提交的比对产品。
5.2.3.3.7注意事项
※预提交 (Pre-Submission):FDA建 议申请者在正式提交De Novo请求前进行预提交,以获得反馈并优化申请。
※时间限制:如果在收到附加信息函后180天内未提交完整回复,申请将被视为撤回。
5.2.4质量体系要求
※自2024年起 ,FDA引入ISO 13485:2016 作为医疗器械质量管理体系的标准,企业需在2026年2月2日前完成过渡。
※企业需建立符合 FDA 21 CFR Part 820 的质量体系,涵盖设计控制、生产、质量控制等环节。
5.2.5临床试验(如适用)
※对于需要临床数据支持的510(k)或PMA申请,企业需获得FDA的器械临床研究豁免(IDE)批准。
※临床研究需符合良好临床实践(GCP)和伦理要求。
5.2.6上市后监管
※产品获批后,企业需遵守FDA的上市后监管要求,包括不良事件报告、定期审查等。
※所有医疗器械需具备UDI(唯一标识符),以便于市场追踪和召回。
06
美国药械大健康市场的采购体系
6.1
药品采购体系
6.1.1集中采购组织(GPO)
6.1.1.1定义与背景
药品集中采购组织 (Group Purchasing Organization) 是一种通过集中医疗机构的采购需求,利用集体购买力与供应商协商折扣的第三方组织。GPO起源于20世纪中期,主要目的是通过集中采购帮助医疗机构降低采购成本。其服务对象包括医院、诊所、疗养院等医疗机构。
6.1.1.2组成结构
※整合医疗网络型GPO:通过控股或正式协议联合一定区域内的医疗机构,享有一致的目标与使命。
※自由GPO:未加入医院联盟的医疗机构自愿加入GPO,参与集团采购,单个医疗机构保持独立性。
6.1.1.3运作模式
6.1.1.3.1需求汇总与合同谈判:
GPO通过调研会员医院的需求,汇总采购量后与供应商进行谈判。GPO以会员的名义与供应商签订合同,但自身并不直接采购产品。通过集中采购,GPO能够为会员医院争取到更优惠的价格和更好的服务条款。
6.1.1.3.2采购流程:
GPO的基本采购流程包括需求调研、发布招标公告(RFP)、与供应商谈判、签订合同等。合同周期通常为3-5年,以确保采购的稳定性和规模经济。
6.1.1.3.3成员与服务:
美国约96%-98%的医院至少加入了一个GPO,平均每家医院加入2-4个GPO。GPO的采购范围不仅包括药品,还涵盖医疗器械、耗材、办公用品、膳食等几乎医院使用的所有产品。
6.1.1.3.4盈利模式:
GPO通常从供应商处收取管理费,一般不超过采购金额的3%。
6.1.2药品福利管理机构(PBM)
6.1.2.1定义与背景
药品福利管理 (Pharmacy Benefit Management) 是一种专业的第三方服务模式,主要通过协调医保支付方、药企、医院、药店和患者等多方利益,实现药品费用的有效管理和医疗服务的优化。PBM起源于20世纪60年代的美国,是基于管理式医疗体系而发展起来的药品价格约束机制。其核心目的是帮助医保支付方(如雇主、商业保险公司和政府)管理日益复杂和昂贵的处方药物使用,同时保障患者获得高质量的医疗服务。
6.1.2.2主要功能
6.1.2.2.1药品目录管理:
PBM通过制定药品目录(Formulary),确定哪些药品可以报销,并根据药品的成本、安全性和疗效进行分类管理。
6.1.2.2.2处方审核与管理:
PBM利用专业的处方审核系统,对处方的合理性和费用进行审核,确保处方符合医保支付方的要求。
6.1.2.2.3药品采购与价格谈判:
PBM凭借其大规模采购的优势,与药企进行价格谈判,争取更低的药品价格,从而降低医保支付方的成本。
6.1.2.2.4用药指导与疾病管理:
PBM通过数据分析和临床路径管理,为患者提供用药指导,优化治疗方案,同时帮助管理慢性疾病。
6.1.2.3运作流程
6.1.2.3.1药品目录制定:
PBM根据药品的疗效、价格和市场需求,制定药品目录,并与保险公司协商确定报销范围。
6.1.2.3.2价格谈判:
PBM与药企进行价格谈判,获取药品的折扣和返点。这些折扣通常会部分让利给患者,以降低其自付费用。
6.1.2.3.3处方审核:
当患者持处方到药房购药时,药房会将处方信息提交给PBM进行审核。PBM会根据药品目录和患者保险信息,确定药品的报销额度。
6.1.2.3.4药品配送:
审核通过后,PBM会安排药品配送到药房,患者只需支付自付部分(Co-pay),剩余费用由PBM与保险公司结算。
6.1.2.3.5数据分析与反馈:
PBM通过数据分析,为保险公司和医疗机构提供用药趋势和成本控制的建议,帮助优化药品管理。
6.1.2.4收入模型
6.1.2.4.1药品折价与返点:PBM通过与药企谈判获得的药品折价(通常为3%)和返点(通常为2%)。
6.1.2.4.2会员费:雇主或患者会员支付的会员费(通常为1%)。
6.1.2.4.3信息化服务收入:PBM通过提供药品目录管理、处方审核和数据分析等信息化服务获取收入
6.1.3直接采购
部分医疗机构(如大型医院或连锁机构)会直接与药品供应商或经销商联系,进行自主采购。这种模式通常用于特定药品或紧急需求,但采购成本可能高于通过GPO采购。
6.2
医疗器械采购体系
6.2.1GPO采购
医疗器械的采购同样主要通过GPO完成。GPO通过集中采购帮助医疗机构降低采购成本,平均为医院节省10%~18%的费用。其采购流程与药品类似,包括需求汇总、供应商投标、评估与谈判、合同签订和发货。
6.2.2医院集团(IDN)采购
IDN是医疗机构横向整合的结果,某些医院没有加入GPO组织,但通过形成联盟的方式共享资源,并由“盟主”与供应商进行价格谈判。IDN在重大采购时可能会与GPO合作。
6.3
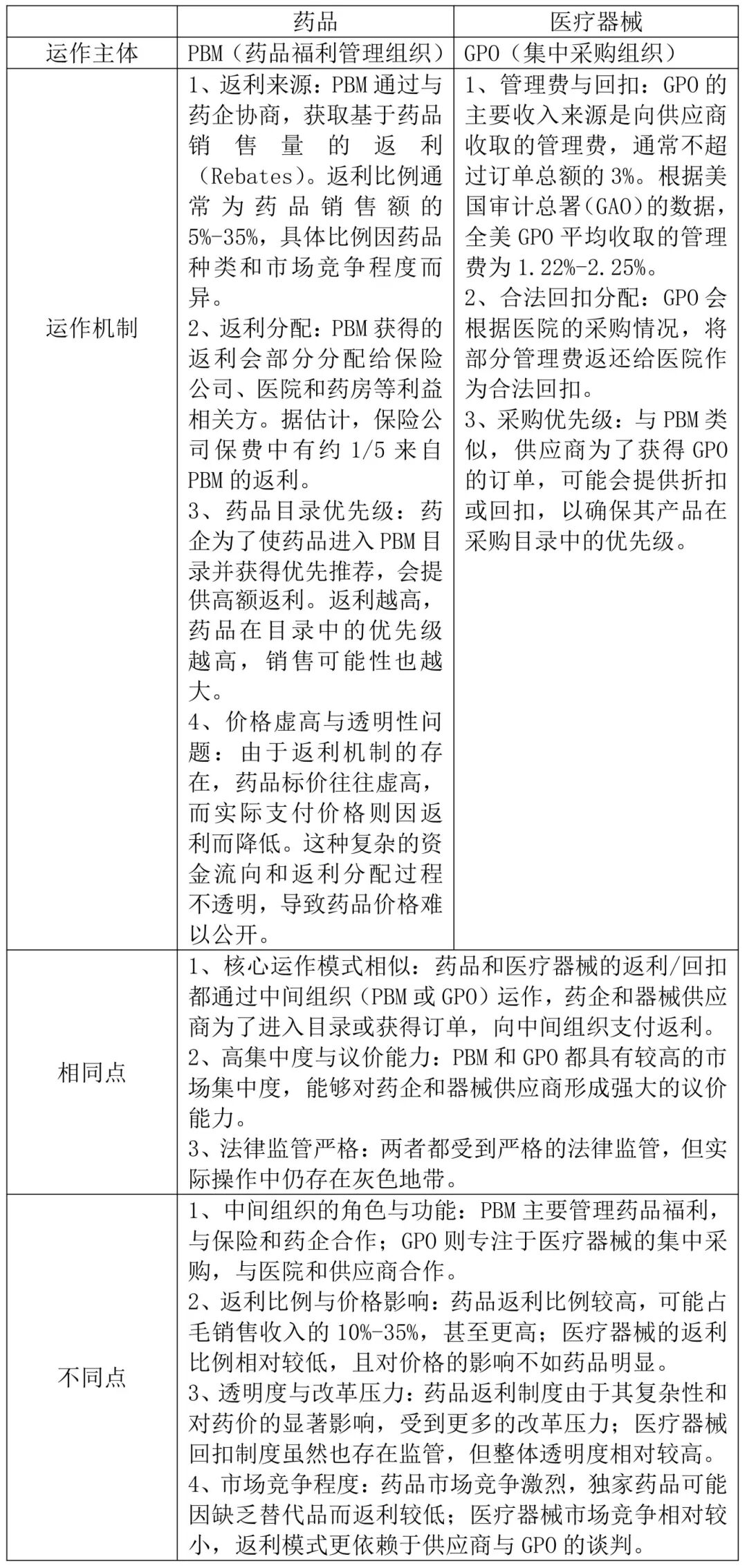
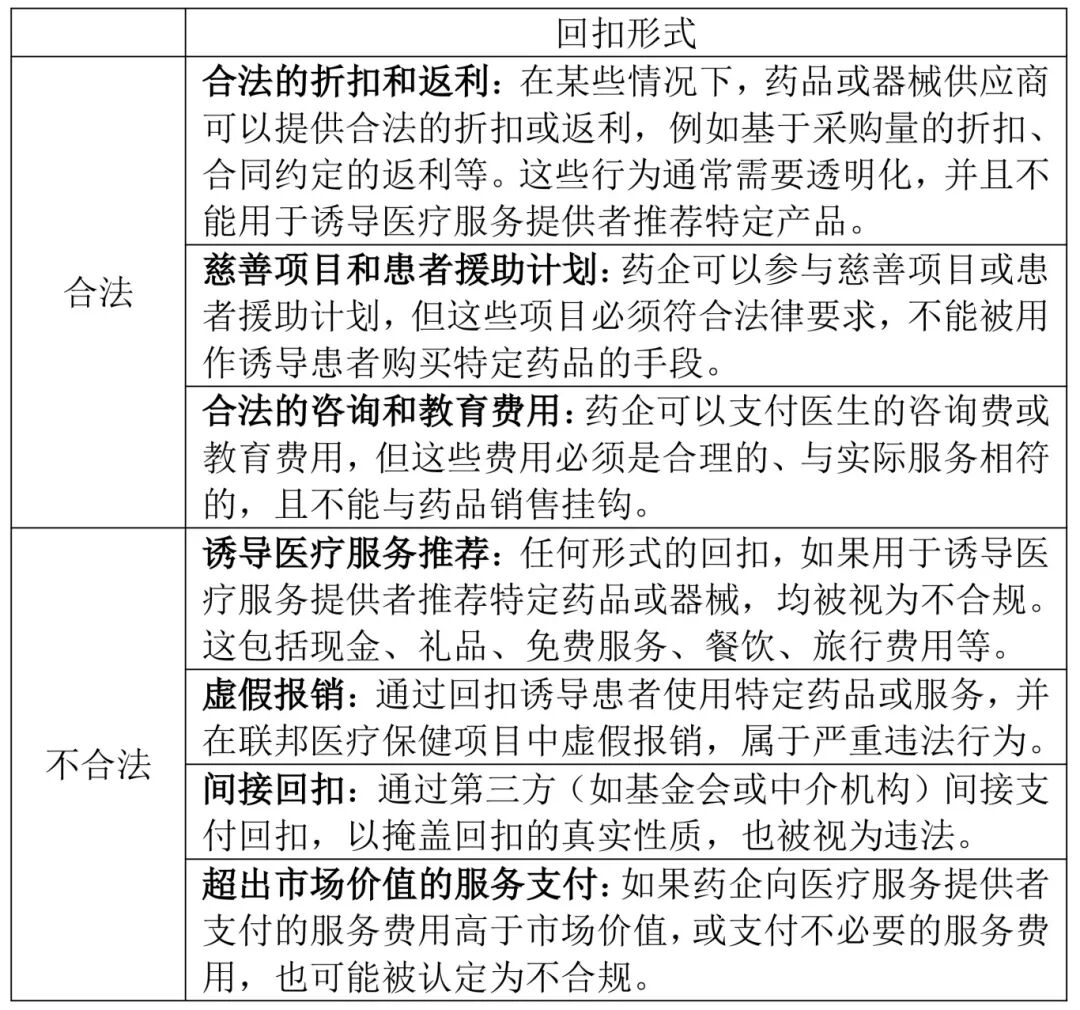
采购中的返利与回扣机制
6.4
回扣机制的法律监管体系
美国的反回扣法律框架主要由以下几部联邦法律构成:
6.4.1《反回扣法》 (Anti-Kickback Statute, AKS)
该法律明确禁止在联邦资助的医疗保健项目中提供、支付、索取或接受任何形式的回扣或其他非法利益,以诱导或回报医疗服务的推荐、安排或使用。回扣的定义非常广泛,包括现金、免费服务、礼品、餐饮、旅行费用等。
6.4.2《斯塔克法》 (Stark Law)
该法律禁止医生在自我推荐医疗服务时收取回扣,覆盖范围包括临床实验室服务、物理治疗、放射学服务、耐用医疗设备等。这是一项严格责任法规,即不需要证明医生有违法意图,只要存在回扣行为即视为违法。
6.4.3《虚假申报法》 (False Claims Act, FCA)
该法律禁止在联邦医疗保健项目中提交虚假报销申请,包括通过回扣诱导患者使用特定药品或服务。
6.4.4 《消除康复产业回扣法案》 (Eliminating Kickbacks in Recovery Act, EKRA)
该法律主要针对康复和实验室医疗保健行业,禁止在这些领域内存在不合规的转诊和支付关系。
07
美国药械大健康市场的销售体系
7.1
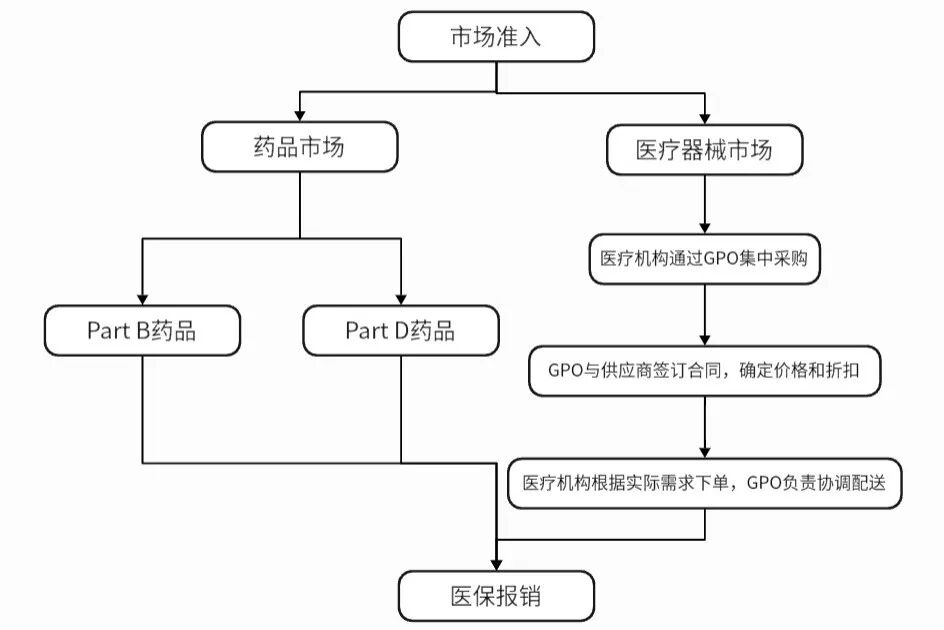
药品销售的市场结构
药品销售主要分为Part B和Part D两个市场:
7.1.1Part B药品:通常指在医院或诊所使用的大分子药物(如抗体药物)。其销售模式为“Buy-and-Bill”,即医院或诊所先购买药品并储存,然后为患者提供服务后向医保局(CMS)提交报销申请。
7.1.2Part D药品:主要涵盖口服药和患者自行注射的药物,由PBM管理。PBM负责药品目录管理、价格谈判和报销申请处理。
7.2
医保报销

7.3
销售流程
08
结语
美国药械大健康市场机遇与挑战并存。其庞大的市场规模、持续增长的趋势以及政府的支持政策,为全球药械大健康企业提供了广阔的发展空间,成为企业拓展国际业务、实现创新突破的重要舞台。
然而,企业在进入和运营过程中,必须高度重视其复杂的法律监管体系。从市场准入阶段各类严格的申请审批要求,到采购、销售环节中对回扣等行为的严格规范,每一项法律规定都紧密关联着企业的运营与发展。稍有不慎,企业就可能面临法律风险,不仅会造成经济损失,还可能影响企业声誉,阻碍未来的市场拓展。
对于出海美国的药械大健康企业而言,深入理解并严格遵守美国的法律法规是成功的关键。企业需要组建专业的法律合规团队,或借助专业的法律机构,提前做好充分的法律准备。在产品研发、临床试验、市场推广等各个环节,都要确保符合美国的法律标准。同时,密切关注美国法律政策的动态变化,及时调整企业战略和运营模式,以适应不断变化的市场环境。
只有这样,企业才能在这片充满潜力的市场中稳健发展,充分把握机遇,实现自身的可持续发展,在国际医药领域中占据一席之地,为全球药械大健康事业的发展贡献力量。
参考文献
[1]“FDA's Center for Drug Evaluation and Research.” U.S. Food and Drug Administration, https://www.fda.gov/drugs/development-approval-process-drugs/how-drugs-are-developed-and-approved.
[2]“21 CFR Chapter I -- Food and Drug Administration, Department of Health and Human Services.” Electronic Code of Federal Regulations, https://www.ecfr.gov/current/title-21/chapter-I.
或许您还想看
作者简介

桂 辛
联席合伙人
桂辛律师是北京德和衡(上海)律师事务所金融投资并购部副主任、上海市律师协会医药健康专业委员会委员、上海政法学院环境资源与能源法研究中心客座教授和兼职硕士生导师、上海涉外高端律师研究生暑期学校授课导师、威科先行入库专家等。桂辛律师荣获法律界多项荣誉,上榜2025新则新势力榜单“律师新势力(40 under 40)”、律新社《精品法律服务品牌指南(2024):生物医药及医疗领域》“匠心律师”等。
桂辛律师致力于成为 “科创家私人/公司法律顾问”,深耕“专精特新”领域多年,尤其在医药与医疗大健康领域有着丰富的实战经验,为各类生物医药、医疗技术及其他大健康企业的研发、临床、生产、商业化、企业合规、数据保护、许可交易等方面提供法律服务。
桂辛律师具有中国和美国双重法学教育背景,国内法学院本科毕业时获得上海市优秀毕业生荣誉,国外法学院硕士毕业时获得美国南加州大学法学院优秀毕业生荣誉且姓名被永久刻在法学院荣誉墙上。另外桂辛律师正在中国人民大学攻读经济学硕士学位。
桂辛律师同时具有公司法务及执业律师复合法律实践经验,除执业律师经验外,桂辛律师曾先后在欧洲上市公司、美国上市公司、中国上市公司的法务部为所在公司提供法律服务并获得优秀员工荣誉。桂辛律师参与过初创企业早期搭建到上市的全过程,也曾负责企业股权转让、许可交易全流程法律服务,同时深度参与企业日常法律事务(包括但不限于公司全业务条线的合同审阅、法律咨询、法律培训等),并协助企业进行战略规划以及合规制度的梳理和建立,为企业重大经营活动提供法律意见和评估。桂辛律师对上市企业内部运营和决策制定以及初创公司/中小型企业的关注和需求有深刻的了解和认识,力求在提供专业法律服务时能够贴合企业实际、为企业的经营及项目开展保驾护航。
桂辛律师还牵头成立了天牧生命科学法律团队,专注于生命科学领域,团队特色和优势是为客户提供“法财税商管”全维度一站式综合服务。团队成员均毕业于国内外知名大学,具有综合教育背景,包括法学、生物制药、经济学、电子信息工程等,工作语言包括英语、日语、韩语、法语、德语、西班牙语、俄语等,持有多项专业资格,包括中国执业律师、中国注册会计师(CICPA)、中国注册税务师(CTA)、美国注册管理会计师(CMA)、美国金融分析师(CFA)、美国金融风险管理师(FRM)、期货投资分析师、微软MTA(Python数据分析)、中国专利代理师等。团队服务过的客户包括但不限于阿斯利康(AstraZeneca)、艾力斯医药、渤健(Biogen)、迪福润丝生物、鼎康生物、复星医药、海尔集团、华润生命科学集团、惠氏制药(Wyeth)、君实生物、兰升生物、丽珠医药、绿叶制药集团、萌蒂制药、美狮生物(Puma Biotechnology, Inc)、诺和诺德(Novo Nordisk)、诺瓦地克(Novaliq)、齐鲁制药、青峰医药、奇华顿、人福医药、睿智医药、石药集团、泰诺麦博、天境生物、易慕峰、远东宏信、悦康科创、中化集团等。
手机:18817328986
邮箱:guixin@deheheng.com
质控人简介

陈焘
高级权益合伙人
金融业务中心总监
END







